
-
国内仿制药注册申报全过程
康安医药咨询
在国内医药环境发展的多年来,已经可以在国内进行仿制药的生产。但是是需要经过非常严格的手续,而且也不是任何仿制药都可以生产。所以很多新的药企需要进行医药生产,所以就需要相关的信息。在此将信息在此提供,以便大家查阅。
在国内医药环境发展的多年来,已经可以在国内进行仿制药的生产。但是是需要经过非常严格的手续,而且也不是任何仿制药都可以生产。所以很多新的药企需要进行医药生产,所以就需要相关的信息。在此将信息在此提供,以便大家查阅。
法规概述
依据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施条例》和《药品注册管理办法》等法律法规,仿制药药品上市前,药品申请人应按要求向国家药品监督管理局药品审评中心(CDE)提出相关药品注册申请,经CDE受理并完成技术审评后转国家药品监督管理局(NMPA)决定是否批准。
企业义务及应对策略
申请人依法向国家药品监督管理局药品审评中心(CDE)提出进口或国产仿制药注册申请。
进口或国产仿制药,境内申请人是在中华人民共和国境内合法登记并能独立承担相应法律责任的企业或者药品研制机构等;境外申请人应当是境外合法制药厂商,境外申请人应当指定中国境内的企业法人办理相关药品注册事项。
注册目标:获取《进口药品注册证》/《医药产品注册证》或药品批准文号。申请人取得药品注册证书后,为药品上市许可持有人。
注册登记及流程
注册类型
进口或国产仿制药,药品注册按照中药、化学药和生物制品等进行分类注册管理。
注册资料要求
中药应符合《国家药监局关于发布中药注册分类及申报资料要求的通告》(2020年第68号)中的要求。
化学药应符合《国家药监局关于发布化学药品注册分类及申报资料要求的通告》(2020年第44号)中的要求。
生物制品应符合《国家药监局关于发布生物制品注册分类及申报资料要求的通告》(2020年第43号)中的要求。
注册流程
国产仿制药申请流程
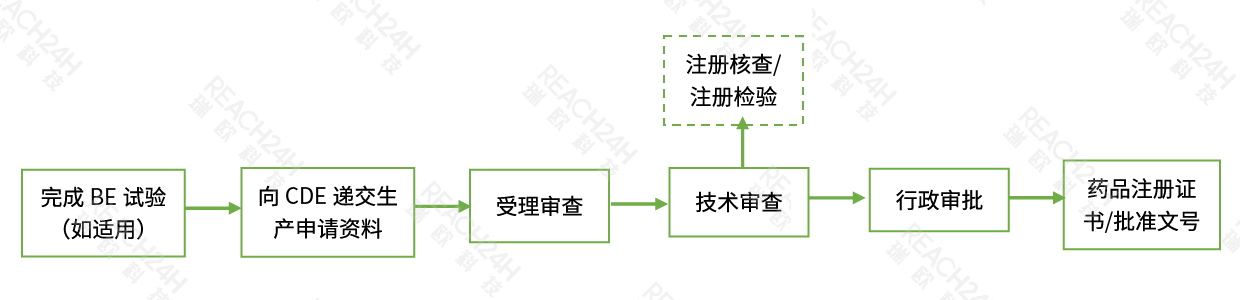
注:根据技术审评需要进行现场核查和注册检验
进口仿制药申请流程
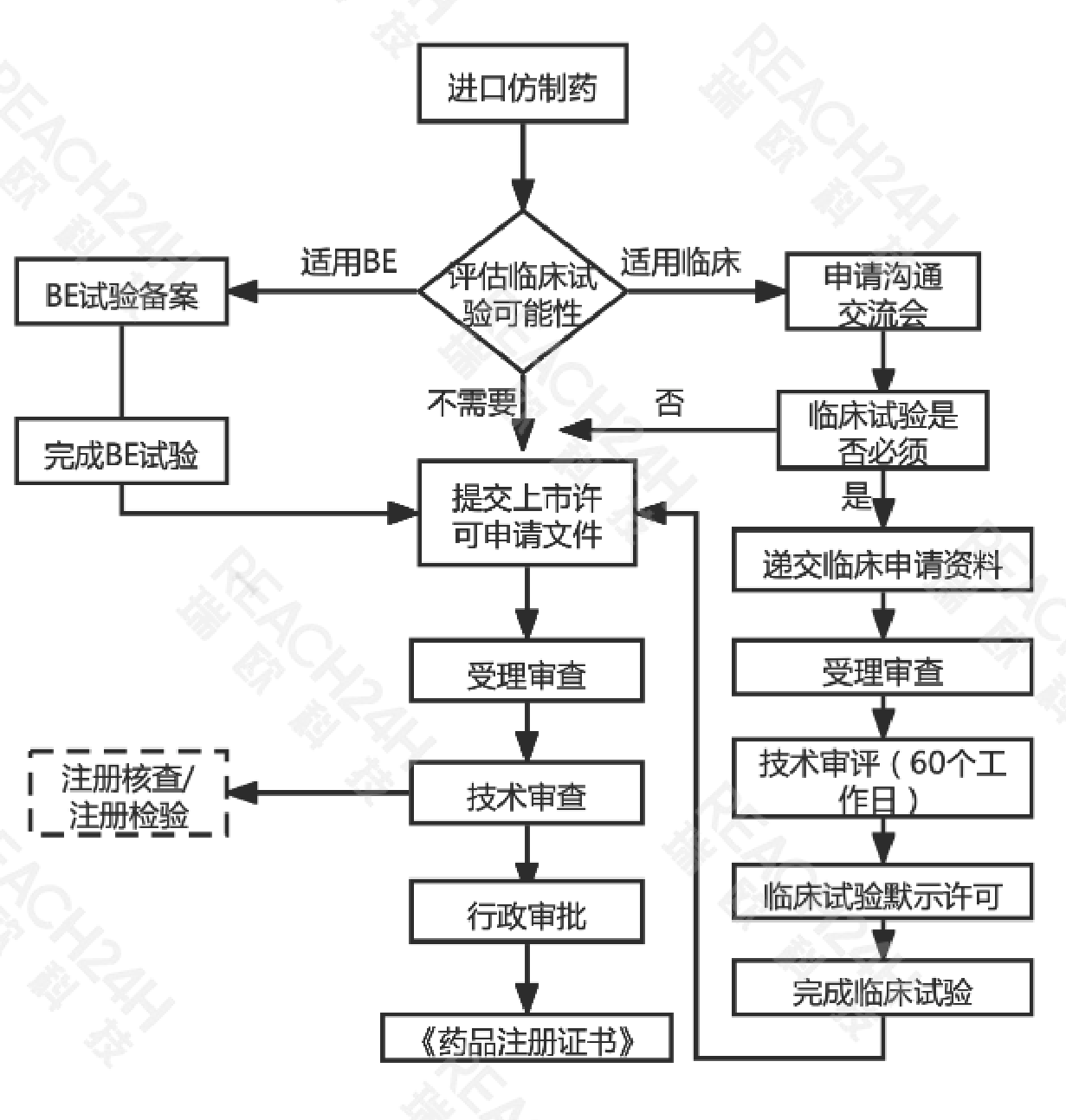
注:根据技术审评需要进行现场核查和注册检验
导航

